 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Phosphodiesterase Inhibitors
General Pharmacology of cAMP-Dependent Phosphodiesterase Inhibitors (PDE3)
Heart
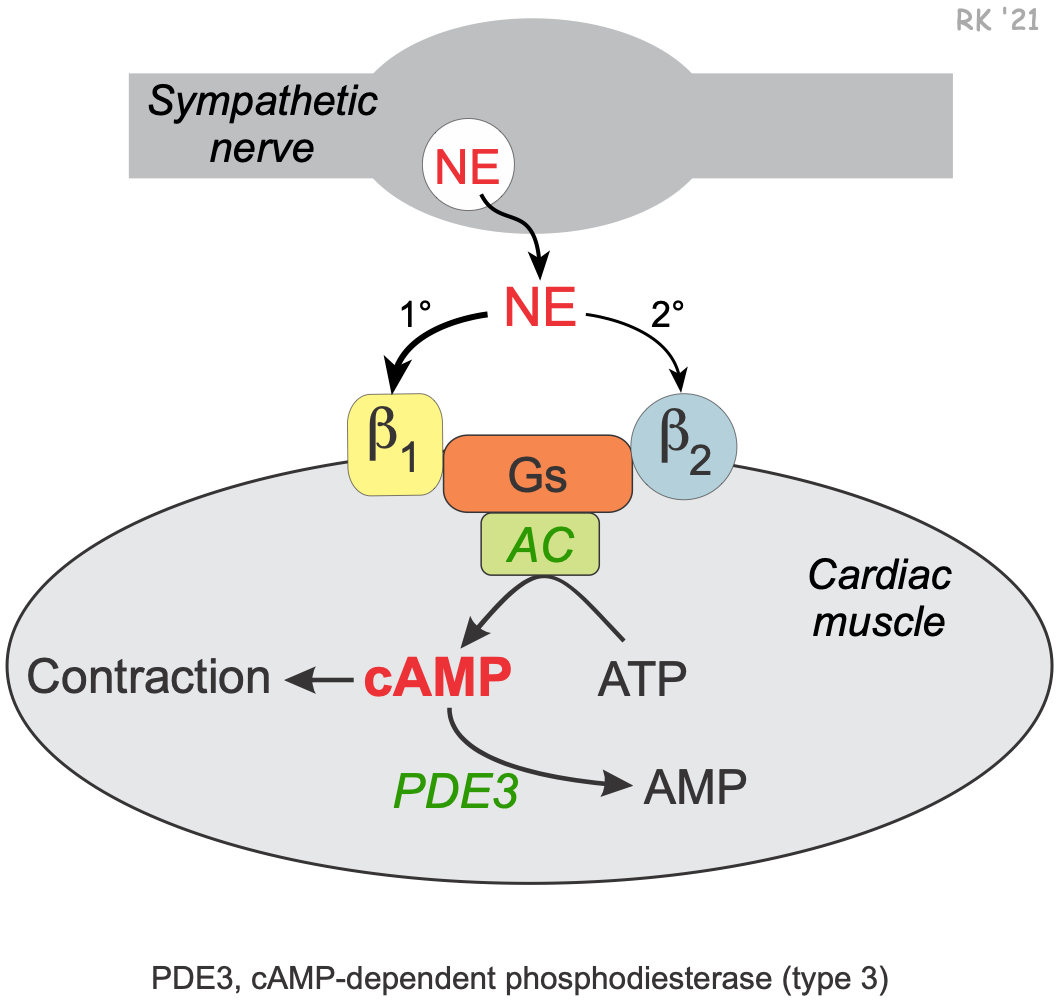 Intracellular concentrations of cAMP play an important second messenger role in regulating cardiac muscle contraction. Activation of the sympathetic nervous system releases the neurotransmitter norepinephrine and increases circulating catecholamines (epinephrine and norepinephrine). These catecholamines bind primarily to beta1-adrenoceptors in the heart that are coupled to Gs-proteins. This activates adenylyl cyclase to form cAMP from ATP. Increased cAMP, through its coupling with other intracellular messengers, increases contractility (inotropy), heart rate (chronotropy) and conduction velocity (dromotropy). Cyclic-AMP is broken down by an enzyme called cAMP-dependent phosphodiesterase (PDE). The isoform of this enzyme that is targeted by currently used clinical drugs is the type 3 form (PDE3). Inhibition of this enzyme prevents cAMP breakdown and increases its intracellular concentration. This increases cardiac inotropy, chronotropy and dromotropy. PDE3 inhibitors can be thought of as a backdoor approach to cardiac stimulation, whereas β-agonists go through the front door to produce the same cardiac effects.
Intracellular concentrations of cAMP play an important second messenger role in regulating cardiac muscle contraction. Activation of the sympathetic nervous system releases the neurotransmitter norepinephrine and increases circulating catecholamines (epinephrine and norepinephrine). These catecholamines bind primarily to beta1-adrenoceptors in the heart that are coupled to Gs-proteins. This activates adenylyl cyclase to form cAMP from ATP. Increased cAMP, through its coupling with other intracellular messengers, increases contractility (inotropy), heart rate (chronotropy) and conduction velocity (dromotropy). Cyclic-AMP is broken down by an enzyme called cAMP-dependent phosphodiesterase (PDE). The isoform of this enzyme that is targeted by currently used clinical drugs is the type 3 form (PDE3). Inhibition of this enzyme prevents cAMP breakdown and increases its intracellular concentration. This increases cardiac inotropy, chronotropy and dromotropy. PDE3 inhibitors can be thought of as a backdoor approach to cardiac stimulation, whereas β-agonists go through the front door to produce the same cardiac effects.
Blood vessels
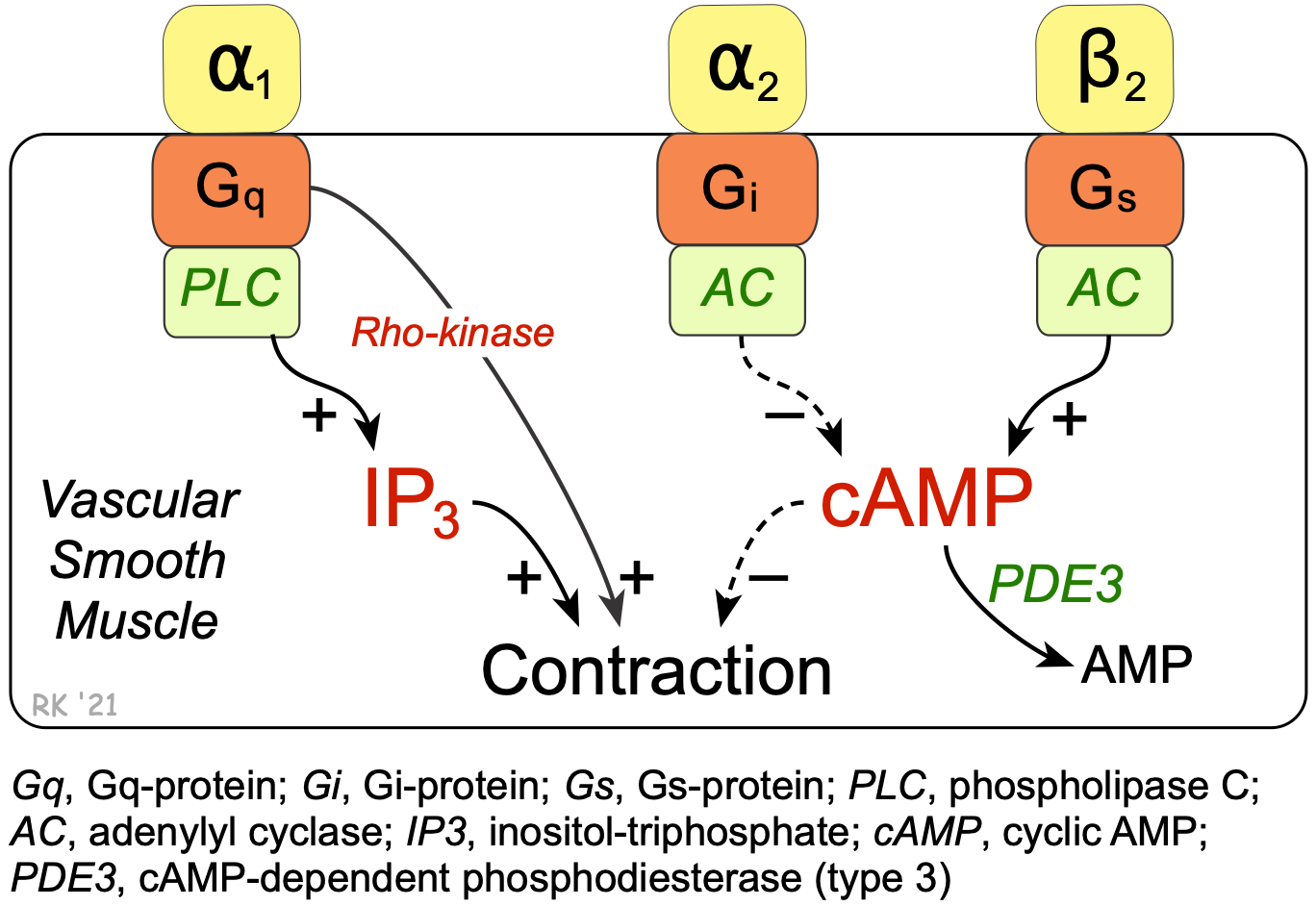 Cyclic-AMP also plays an important role in regulating the contraction of vascular smooth muscle. Beta2-adrenoceptor agonists such as epinephrine stimulate the Gs-protein and the formation of cAMP (click here for details). Unlike cardiac muscle, increased cAMP in smooth muscle causes relaxation. This is because cAMP normally inhibits myosin light chain kinase, the enzyme that phosphorylates smooth muscle myosin, causing contraction. Like the heart, the cAMP is broken down by a cAMP-dependent PDE (PDE3). Therefore, inhibition of this enzyme increases intracellular cAMP, which further inhibits myosin light chain kinase, producing less contractile force (i.e., promoting relaxation).
Cyclic-AMP also plays an important role in regulating the contraction of vascular smooth muscle. Beta2-adrenoceptor agonists such as epinephrine stimulate the Gs-protein and the formation of cAMP (click here for details). Unlike cardiac muscle, increased cAMP in smooth muscle causes relaxation. This is because cAMP normally inhibits myosin light chain kinase, the enzyme that phosphorylates smooth muscle myosin, causing contraction. Like the heart, the cAMP is broken down by a cAMP-dependent PDE (PDE3). Therefore, inhibition of this enzyme increases intracellular cAMP, which further inhibits myosin light chain kinase, producing less contractile force (i.e., promoting relaxation).
Overall cardiovascular effects
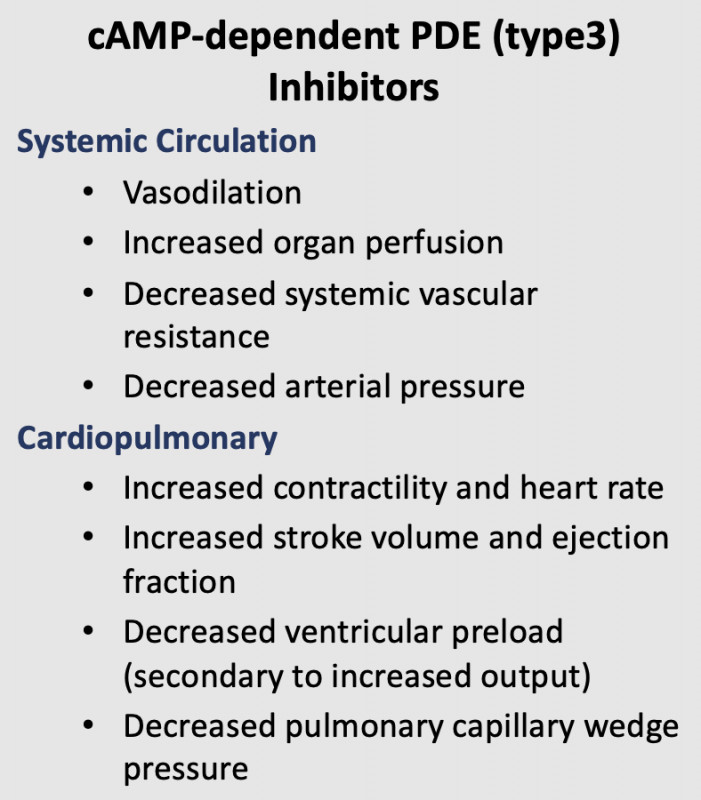 The cardiac and vascular effects of cAMP-dependent PDE inhibitors cause cardiac stimulation, which increases cardiac output, and reduced systemic vascular resistance, which lowers arterial pressure. Because cardiac output increases and systemic vascular resistance decreases, the change in arterial pressure depends on the relative effects of the PDE inhibitor on the heart versus the vasculature. At normal therapeutic doses, PDE3 inhibitors such as milrinone have a greater vascular than cardiac effect, so that arterial pressure is lowered in the presence of augmented cardiac output. Because of the dual cardiac and vascular effects of these compounds, they are sometimes referred to as "inodilators."
The cardiac and vascular effects of cAMP-dependent PDE inhibitors cause cardiac stimulation, which increases cardiac output, and reduced systemic vascular resistance, which lowers arterial pressure. Because cardiac output increases and systemic vascular resistance decreases, the change in arterial pressure depends on the relative effects of the PDE inhibitor on the heart versus the vasculature. At normal therapeutic doses, PDE3 inhibitors such as milrinone have a greater vascular than cardiac effect, so that arterial pressure is lowered in the presence of augmented cardiac output. Because of the dual cardiac and vascular effects of these compounds, they are sometimes referred to as "inodilators."
Other actions
PDE3 inhibitors also decrease platelet aggregation by increasing platelet cAMP. However, only cilostazol (see below) is used for this purpose in the treatment of intermittent claudication (ischemic leg pain associated with leg movement).
General Pharmacology of cGMP-Dependent Phosphodiesterase Inhibitors (PDE5)
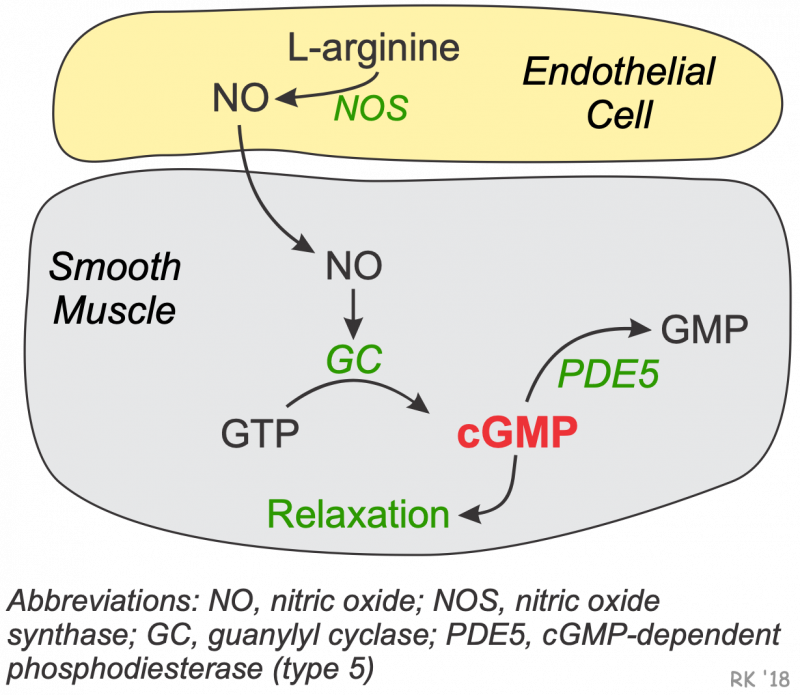 There is a second isoenzyme form of PDE in vascular smooth muscle that is a cGMP-dependent phosphodiesterase. The type 5 isoform of this enzyme (PDE5) is found in the corpus cavernosum of the penis and in vascular smooth muscle. This enzyme breaks down cGMP that forms in response to increased nitric oxide (NO). Increased intracellular cGMP inhibits calcium entry into the cell, decreasing intracellular calcium concentrations and causing smooth muscle relaxation (click here for details).
There is a second isoenzyme form of PDE in vascular smooth muscle that is a cGMP-dependent phosphodiesterase. The type 5 isoform of this enzyme (PDE5) is found in the corpus cavernosum of the penis and in vascular smooth muscle. This enzyme breaks down cGMP that forms in response to increased nitric oxide (NO). Increased intracellular cGMP inhibits calcium entry into the cell, decreasing intracellular calcium concentrations and causing smooth muscle relaxation (click here for details).
NO also activates K+ channels, which leads to hyperpolarization and relaxation. Finally, NO acting through cGMP can stimulate a cGMP-dependent protein kinase that activates myosin light chain phosphatase, the enzyme that dephosphorylates myosin light chains, which leads to relaxation. Therefore, inhibitors cGMP-dependent phosphodiesterase, by increasing intracellular cGMP, enhance smooth muscle relaxation and vasodilation, and cause penile erection.
Therapeutic Indications
The cardiostimulatory and vasodilatory actions of PDE3 inhibitors make them suitable for treating heart failure. Arterial dilation reduces afterload on the failing ventricle and leads to an increase in stroke volume and ejection fraction, as well as increases organ perfusion. Reducing the afterload leads to a secondary decrease in preload on the heart that helps to improve the mechanical efficiency of dilated hearts and to reduce ventricular wall stress and the oxygen demands placed on the failing heart. The cardiostimulatory effects of these drugs increase inotropy, which further enhances stroke volume and ejection fraction. Tachycardia, however, also results, and this is not beneficial; therefore, doses are used that minimize the positive chronotropic actions of the drug. A baroreceptor reflex, which occurs in response to hypotension, may contribute to tachycardia. Clinical trials have shown that long-term therapy with PDE3 inhibitors increases mortality in heart failure patients; therefore, these drugs are not used for long-term, chronic therapy. They are very useful, however, in treating acute, decompensated heart failure or temporary bouts of decompensated chronic failure. They are not used as monotherapy. Instead, they are used with other treatment modalities such as diuretics, ACE inhibitors, beta-blockers or digitalis.
The somewhat selective vasodilatory actions of PDE5 inhibitors have made these compounds very useful in the treatment of male erectile dysfunction. The PDE5 inhibitor sildenafil is also approved to treat pulmonary hypertension.
Specific Drugs
Several PDE inhibitors are available for clinical use:
- PDE3 inhibitors
- milrinone
- inamrinone (formerly amrinone)
- cilostazol
- PDE5 inhibitors
- sildenafil
- tadalafil
The PDE3 inhibitors (except cilostazol) are used for treating acute, decompensated heart failure, whereas the PDE5 inhibitors are used for treating male erectile dysfunction and pulmonary hypertension. Note that the PDE3 inhibitors used in acute heart failure end in "one," whereas the PDE5 inhibitors end in "fil".
Inhibition of platelet aggregation, along with vasodilation, is an important mechanism of action for cilostazol, which is used in the treatment of intermittent claudication in peripheral arterial disease. Cilostazol appears to have less cardiostimulatory effects than milrinone.
Side Effects and Contraindications
PDE3 inhibitors
Milrinone and inamrinone are not used in the treatment of chronic heart failure because clinical trials have shown that long-term use of these drugs worsen patient outcomes. The most common and severe side effect of PDE3 inhibitors is ventricular arrhythmias in about 12% of patients, some of which may be life-threatening. Headaches and hypotension occur in about 3% of patients. These side effects are not uncommon for drugs that increase cAMP in cardiac and vascular tissues, other examples being β-agonists.
PDE5 inhibitors
The most common side effects for PDE5 inhibitors include headache and cutaneous flushing, both of which are related to vascular dilation caused by increased vascular cGMP. There is clinical evidence that nitrodilators may interact adversely with PDE5 inhibitors. The reason for this adverse reaction is that nitrodilators stimulate cGMP production, while PDE5 inhibitors inhibit cGMP degradation. When combined, these two drug classes potentiate cGMP levels, which can lead to hypotension and impaired coronary perfusion.
Revised 02/01/2024