 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Cardiac Glycosides (Digoxin)
Cardiac glycosides represent a family of compounds that are derived from the foxglove plant (Digitalis purpurea). The therapeutic benefits of digitalis were first described by William Withering in 1785. Initially, digitalis was used to treat dropsy, which is an old term for edema. Subsequent investigations found that digitalis was most useful for edema that was caused by a weakened heart (i.e., heart failure).
Mechanism of action
Digitalis compounds are potent inhibitors of cellular Na+/K+-ATPase. This ion transport system moves sodium ions out of the cell and brings potassium ions into the cell. This transport function is necessary for cell survival because sodium diffusion into the cell and potassium diffusion out of the cell down their concentration gradients would reduce their concentration differences (gradients) across the cell membrane over time. Loss of these ion gradients would lead to cellular depolarization and loss of the negative membrane potential that is required for normal cell function. The Na+/K+-ATPase also plays an active role in membrane potential generation. This pump generates electrogenic hyperpolarizing currents because it transports 3 sodium ions out of the cell for every two potassium ions that enter the cell. This can add several negative millivolts to the membrane potential, depending on the activity of the pump.
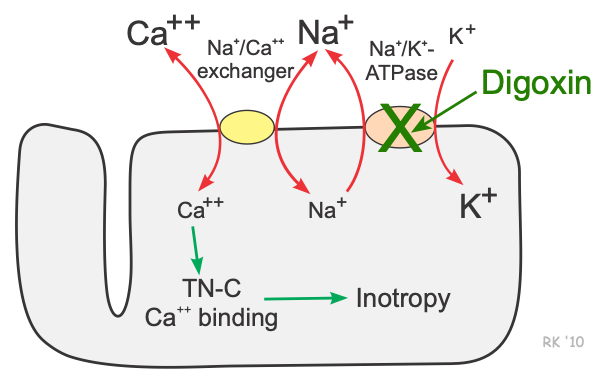 Cardiac myocytes, as well as many other cells, have a Na+-Ca++ exchanger (not an active energy-requiring pump) that is essential for maintaining sodium and calcium homeostasis. Utilizing this exchange pump, calcium and sodium can move in either direction across the sarcolemma. Furthermore, three sodium ions are exchanged for each calcium, therefore an electrogenic potential is generated by this exchanger. The direction of movement of these ions (either inward or outward) depends upon the membrane potential and the chemical gradient for the ions. At rest, the exchanger contributes a few millivolts of depolarizing current because 3 Na+ ions enter the cell for each Ca++ ion that leaves the cell. An increase in intracellular sodium concentration competes for calcium through this exchange mechanism, leading to an increase in intracellular calcium concentration. As intracellular sodium increases, the concentration gradient driving sodium into the cell across the exchanger is reduced, thereby reducing the activity of the exchanger, which decreases the movement of calcium out of the cell. Therefore, mechanisms that lead to an accumulation of intracellular sodium cause a subsequent accumulation of intracellular calcium because of decreased exchange pump activity.
Cardiac myocytes, as well as many other cells, have a Na+-Ca++ exchanger (not an active energy-requiring pump) that is essential for maintaining sodium and calcium homeostasis. Utilizing this exchange pump, calcium and sodium can move in either direction across the sarcolemma. Furthermore, three sodium ions are exchanged for each calcium, therefore an electrogenic potential is generated by this exchanger. The direction of movement of these ions (either inward or outward) depends upon the membrane potential and the chemical gradient for the ions. At rest, the exchanger contributes a few millivolts of depolarizing current because 3 Na+ ions enter the cell for each Ca++ ion that leaves the cell. An increase in intracellular sodium concentration competes for calcium through this exchange mechanism, leading to an increase in intracellular calcium concentration. As intracellular sodium increases, the concentration gradient driving sodium into the cell across the exchanger is reduced, thereby reducing the activity of the exchanger, which decreases the movement of calcium out of the cell. Therefore, mechanisms that lead to an accumulation of intracellular sodium cause a subsequent accumulation of intracellular calcium because of decreased exchange pump activity.
By inhibiting the Na+/K+- ATPase, cardiac glycosides such as digoxin cause intracellular sodium concentration to increase. This leads to an accumulation of intracellular calcium via the Na+- Ca++ exchanger. In the heart, increased intracellular calcium causes more calcium to be taken up and subsequently released by the sarcoplasmic reticulum, thereby making more calcium available to bind to troponin-C, which increases contractility (inotropy). Inhibition of the Na+/K+- ATPase in vascular smooth muscle causes depolarization, which causes smooth muscle contraction and vasoconstriction.
By mechanisms that are not fully understood, digitalis compounds also increase vagal efferent activity to the heart. This parasympathomimetic action of digitalis reduces sinoatrial firing rate (decreases heart rate; negative chronotropy) and reduces conduction velocity of electrical impulses through the atrioventricular node (negative dromotropy).
Pharmacokinetics and toxicity
The long half-life of digoxin distinguishes this drug from most other cardiovascular acting drugs. With a half-life of 40 hours, digoxin requires several days of constant dosing to reach steady-state plasma level. Therefore, when initiating treatment, a special dosing regimen involving either oral or intravenous “loading doses” is used to rapidly increase digoxin plasma levels. This process is termed “digitalization.” Digoxin is eliminated by the kidneys.
The therapeutic plasma concentration range for digoxin is 0.5–1.5 ng/ml. It is important that therapeutic plasma levels are not exceeded because digitalis compounds have a narrow therapeutic safety window, meaning that small increases in plasma concentration within the upper therapeutic range and above can have significant adverse side effects. Plasma concentrations above 2.0 ng/ml can lead to digitalis toxicity, which is frequently manifested as arrhythmias, some of which may be life-threatening. If toxicity occurs with digoxin, it may take several days for the plasma concentrations to fall to safe levels because of the long half-life. There is available for digoxin toxicity an immune Fab (Digibind) that can be used to rapidly reduce plasma digoxin levels. Potassium supplementation can also reverse the toxic effects of digoxin if the toxicity is related to hypokalemia (see below).
Drug Interactions
Many commonly used drugs interact with digoxin. The Class IA antiarrhythmic, quinidine, competes with digoxin for binding sites and depresses renal clearance of digoxin. These effects increase digoxin levels and can produce toxicity. Similar interactions occur with calcium-channel blockers and non-steroidal anti-inflammatory drugs. Other drugs that interact with digoxin are amiodarone (Class III antiarrhythmic) and beta-blockers. Diuretics can indirectly interact with digoxin because of their potential for decreasing plasma potassium levels (i.e., producing hypokalemia). Hypokalemia results in increased digoxin binding to the Na+/K+-ATPase and thereby enhances digoxin's therapeutic and toxic effects. Hypercalcemia enhances digitalis-induced increases in intracellular calcium, which can lead to calcium overload and increased susceptibility to digoxin-induced arrhythmias. Hypomagnesemia also sensitizes the heart to digoxin-induced arrhythmias.
Therapeutic Uses
Heart failure
Digoxin has historically been used in the treatment of chronic heart failure because of its cardiotonic effect. Although newer and more efficacious treatments for heart failure are available, digoxin is still used. Clinical studies in heart failure patients with reduced ejection fraction (HFrEF) have shown that digoxin, when used with diuretics and vasodilators, improves cardiac output and ejection fraction. It also reduces filling pressures and pulmonary capillary wedge pressures (this reduces pulmonary congestion and edema); heart rate changes little. These effects are to be expected for a drug that increases inotropy. Although the direct effect of digoxin on blood vessels is vasoconstriction, when given to patients in heart failure, the systemic vascular resistance falls. This likely results from the improvement in cardiac output, which leads to withdrawal of compensatory vasoconstrictor mechanisms (e.g., sympathetic adrenergic activity and angiotensin II influences).
Atrial fibrillation and flutter
Atrial fibrillation and flutter lead to a rapid ventricular rate that can impair ventricular filling (due to decreased filling time) and reduce cardiac output. Furthermore, chronic ventricular tachycardia can lead to heart failure. Digoxin, although not a first-line drug for rate control, can be used to reduce ventricular rate when a high atrial rate or atrial fibrillation is driving it. The mechanism of this beneficial effect of digoxin is its ability to activate vagal efferent nerves to the heart (parasympathomimetic effect). Vagal activation can reduce the conduction of electrical impulses within the atrioventricular node to the point where some impulses will be blocked. When this occurs, fewer impulses reach the ventricles and the ventricular rate falls. Digoxin also increases the effective refractory period within the atrioventricular node.
Side Effects, Contraindications, and Warnings
The major side effect of digoxin is cardiac arrhythmia, especially atrial tachycardias and atrioventricular block. Digoxin is contraindicated in patients who are hypokalemic, or who have atrioventricular block or Wolff-Parkinson-White (WPW) syndrome. Impaired renal function leads to enhanced plasma levels of digoxin because digoxin is eliminated by the kidneys. Lean, elderly patients are more susceptible to digoxin toxicity because they often have reduced renal function. This, along with their reduced muscle mass, increases plasma digoxin levels at a given dose because muscle Na+/K+-ATPase acts as a large binding reservoir for digoxin. A 2018 analysis of the AFFIRM trial determined that digoxin significantly increased all-cause mortality in patients with atrial fibrillation. This calls into question the practice of using digoxin for lowering ventricular rate in patients with atrial fibrillation.
Revised 01/28/2024