 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Diuretics
General Pharmacology
Renal handling of sodium and water
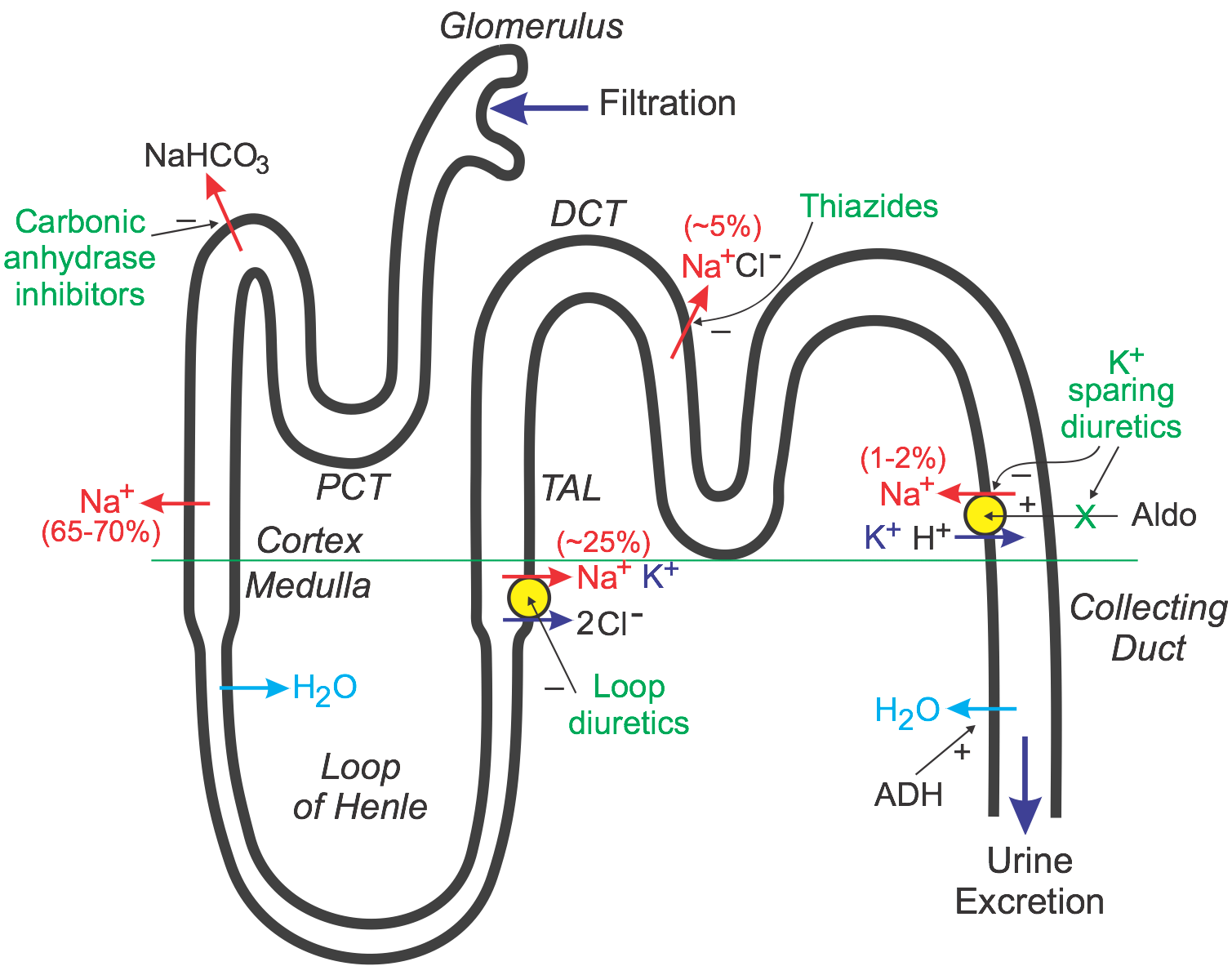 To understand the action of diuretics, it is first necessary to review how the kidney filters fluid and forms urine. The following discussion and accompanying illustration provide a simple overview of how the kidney handles water and electrolytes. For a more detailed explanation, particularly related to ion and fluid movement across the renal tubular cells, the reader should consult a physiology textbook.
To understand the action of diuretics, it is first necessary to review how the kidney filters fluid and forms urine. The following discussion and accompanying illustration provide a simple overview of how the kidney handles water and electrolytes. For a more detailed explanation, particularly related to ion and fluid movement across the renal tubular cells, the reader should consult a physiology textbook.
As blood flows through the kidney, it passes into glomerular capillaries located within the cortex (outer zone of the kidney). These glomerular capillaries are highly permeable to water and electrolytes. Glomerular capillary hydrostatic pressure drives (filters) water and electrolytes into Bowman's space and into the proximal convoluting tubule (PCT). About 20% of the plasma that enters the glomerular capillaries is filtered (termed filtration fraction). The PCT, which lies within the cortex, is the site of sodium, water, and bicarbonate transport from the filtrate (urine), across the tubule wall, and into the interstitium of the cortex. About 65-70% of the filtered sodium is removed from the urine found within the PCT (this is termed sodium reabsorption). This sodium is reabsorbed iso-osmotically, meaning that every molecule of sodium that is reabsorbed is accompanied by a molecule of water. As the tubule dives into the medulla, or middle zone of the kidney, the tubule becomes narrower and forms a loop (Loop of Henle). This loop reenters the cortex as the thick ascending limb (TAL) that travels back to near the glomerulus. Because the interstitium of the medulla is very hyperosmotic and the Loop of Henle is permeable to water, water is reabsorbed from the Loop of Henle and into the medullary interstitium. This loss of water concentrates the urine within the Loop of Henle.
The TAL, which is impermeable to water, has a co-transport system that reabsorbs sodium, potassium, and chloride at a ratio of 1:1:2. Approximately 25% of the sodium load of the original filtrate is reabsorbed at the TAL. From the TAL, the urine flows into the distal convoluting tubule (DCT), which is another site of sodium transport (~5% via a sodium-chloride co-transporter) into the cortical interstitium (the DCT is also impermeable to water). Finally, the tubule dives back into the medulla as the collecting duct and then into the renal pelvis, where it joins with other collecting ducts to exit the kidney as the ureter. The distal segment of the DCT and the upper collecting duct have a transporter that reabsorbs sodium (about 1-2% of filtered load) in exchange for potassium and hydrogen ions, which are excreted into the urine. It is important to note two things about this transporter. First, its activity is dependent on the tubular concentration of sodium, so that when sodium is high, more sodium is reabsorbed, and more potassium and hydrogen ions are excreted. Second, this transporter is regulated by aldosterone, which is a mineralocorticoid hormone secreted by the adrenal cortex. Increased aldosterone stimulates the reabsorption of sodium, which also increases the loss of potassium and hydrogen ions to the urine. Finally, water is reabsorbed in the collecting duct through special pores that are regulated by antidiuretic hormone, which is released by the posterior pituitary. ADH increases the permeability of the collecting duct to water, which leads to increased water reabsorption, more concentrated urine, and reduced urine outflow (antidiuresis). Most of the sodium originally filtered is reabsorbed by the kidney so that less than 1% of the originally filtered sodium remains in the final urine.
Mechanisms of diuretic drugs
Diuretic drugs increase urine output by the kidney (i.e., promote diuresis). This is accomplished by altering how the kidney handles sodium. If the kidney excretes more sodium, then water excretion will also increase. Most diuretics produce diuresis by inhibiting the reabsorption of sodium at different segments of the renal tubular system. Occasionally, a combination of two diuretics is given because this can be significantly more effective than either compound alone (synergistic effect). This is because one nephron segment can compensate for altered sodium reabsorption at another nephron segment; therefore, blocking multiple nephron sites significantly enhances efficacy.
Loop diuretics inhibit the sodium-potassium-chloride co-transporter in the thick ascending limb (see above figure). This transporter normally reabsorbs about 25% of the sodium load; therefore, inhibition of this pump can lead to a significant increase in the distal tubular concentration of sodium, reduced hypertonicity of the surrounding interstitium, and less water reabsorption in the collecting duct. This altered handling of sodium and water leads to both diuresis (increased water loss) and natriuresis (increased sodium loss). By acting on the thick ascending limb, which handles a significant fraction of sodium reabsorption, loop diuretics are powerful diuretics. These drugs also induce renal synthesis of prostaglandins, which contributes to their renal action, including the increase in renal blood flow and redistribution of renal cortical blood flow. Loop diuretics are the most effective diuretic class because their site of action has a high capacity for sodium reabsorption. Note that efficacy is inversely related to renal function, which can be impaired in heart failure.
Thiazide diuretics, which are the most prescribed diuretic, inhibit the sodium-chloride transporter in the distal tubule. Because this transporter normally only reabsorbs about 5% of filtered sodium, these diuretics are less efficacious than loop diuretics in producing diuresis and natriuresis. Nevertheless, they are sufficiently powerful to satisfy many therapeutic needs requiring a diuretic. Their mechanism depends on renal prostaglandin production.
Because loop and thiazide diuretics increase sodium delivery to the distal segment of the distal tubule, this increases potassium loss (potentially causing hypokalemia). This occurs because the increase in distal tubular sodium concentration stimulates the aldosterone-sensitive sodium pump to increase sodium reabsorption in exchange for potassium and hydrogen ions, which are lost to the urine. The increased hydrogen ion loss can lead to metabolic alkalosis. Part of the loss of potassium and hydrogen ions by loop and thiazide diuretics results from activation of the renin-angiotensin-aldosterone system that occurs because of reduced blood volume and arterial pressure. Increased aldosterone stimulates sodium reabsorption and increases potassium and hydrogen ion excretion into the urine.
There is a third class of diuretic that is functionally referred to as potassium-sparing diuretics. Unlike loop and thiazide diuretics, some of these drugs do not act directly on sodium transport. Some drugs in this class antagonize the actions of aldosterone (aldosterone receptor antagonists; mineralocorticoid receptor antagonists; MRAs) at the distal segment of the distal tubule. This causes more sodium (and water) to pass into the collecting duct and be excreted in the urine. They are called K+-sparing diuretics because they do not produce hypokalemia like the loop and thiazide diuretics. This is because by inhibiting aldosterone-sensitive sodium reabsorption, less potassium and hydrogen ions are exchanged for sodium by this transporter, and therefore less potassium and hydrogen are lost to the urine. Other potassium-sparing diuretics directly inhibit sodium channels associated with the aldosterone-sensitive sodium pump and therefore have similar effects on potassium and hydrogen ions as the aldosterone antagonists. Their mechanism depends on renal prostaglandin production. Because this class of diuretic has weak effects on overall sodium balance, they are often used with thiazide or loop diuretics to help prevent hypokalemia.
Carbonic anhydrase inhibitors inhibit the transport of bicarbonate out of the proximal convoluted tubule into the interstitium, which leads to less sodium reabsorption at this site and therefore greater sodium, bicarbonate, and water loss in the urine. These are the weakest of the diuretics and are seldom, if ever, used in cardiovascular disease.
Cardiovascular effects of diuretics
Through their effects on sodium and water balance, diuretics decrease blood volume and venous pressure. This decreases cardiac filling (preload) and, by the Frank-Starling mechanism, decreases ventricular stroke volume and cardiac output, which leads to a fall in arterial pressure. The decrease in venous pressure reduces capillary hydrostatic pressure, which decreases capillary fluid filtration and promotes capillary fluid reabsorption, thereby reducing edema if present. There is some evidence that loop diuretics cause venodilation, which can contribute to the lowering of venous pressure. Long-term use of diuretics results in a fall in systemic vascular resistance (by unknown mechanisms) that helps to sustain the reduction in arterial pressure.
Therapeutic Uses
Hypertension
Most patients with hypertension, of whom 90-95% have hypertension of unknown origin (primary or essential hypertension), are effectively treated with diuretics. Antihypertensive therapy with diuretics is particularly effective when coupled with reduced dietary sodium intake. The efficacy of these drugs is derived from their ability to reduce blood volume, cardiac output, and, with long-term therapy, systemic vascular resistance. Thiazide diuretics, particularly chlorthalidone, are considered “first-line therapy” for stage 1 hypertension. Potassium-sparing, aldosterone-blocking diuretics (e.g., spironolactone or eplerenone) are used in secondary hypertension caused by primary hyperaldosteronism and sometimes as an adjunct to thiazide treatment in primary hypertension to prevent hypokalemia.
Heart failure
Heart failure leads to activation of the renin-angiotensin-aldosterone system, which causes increased sodium and water retention by the kidneys. This in turn increases blood volume and contributes to the elevated venous pressures associated with heart failure, which can lead to pulmonary and systemic edema. The primary use for diuretics in heart failure is to reduce pulmonary and systemic congestion and edema, and associated clinical symptoms (e.g., shortness of breath—dyspnea). Long-term treatment with diuretics may also reduce the afterload on the heart by promoting systemic vasodilation, which can lead to improved ventricular ejection.
 When treating heart failure with diuretics, care must be taken to not unload too much volume because this can depress cardiac output. For example, if pulmonary capillary wedge pressure is 25 mmHg (point A in figure) and pulmonary congestion is present, a diuretic can safely reduce that elevated pressure to a level (e.g., 14 mmHg; point B in figure) that will reduce pulmonary pressures without compromising ventricular stroke volume. This is because heart failure caused by systolic dysfunction is associated with a depressed, flattened Frank-Starling curve. However, if the volume is reduced too much, stroke volume will fall because the heart will now be operating on the ascending limb of the Frank-Starling relationship. If the heart failure is caused by diastolic dysfunction, diuretics must be used cautiously to not impair ventricular filling. In diastolic dysfunction, ventricular filling requires elevated filling pressures because of the reduced ventricular compliance.
When treating heart failure with diuretics, care must be taken to not unload too much volume because this can depress cardiac output. For example, if pulmonary capillary wedge pressure is 25 mmHg (point A in figure) and pulmonary congestion is present, a diuretic can safely reduce that elevated pressure to a level (e.g., 14 mmHg; point B in figure) that will reduce pulmonary pressures without compromising ventricular stroke volume. This is because heart failure caused by systolic dysfunction is associated with a depressed, flattened Frank-Starling curve. However, if the volume is reduced too much, stroke volume will fall because the heart will now be operating on the ascending limb of the Frank-Starling relationship. If the heart failure is caused by diastolic dysfunction, diuretics must be used cautiously to not impair ventricular filling. In diastolic dysfunction, ventricular filling requires elevated filling pressures because of the reduced ventricular compliance.
Most patients in heart failure are prescribed a loop diuretic because they are more effective in unloading sodium and water than thiazide diuretics. In mild heart failure, a thiazide diuretic may be used. Potassium-sparing, aldosterone-blocking diuretics (e.g., spironolactone) are being used increasingly in heart failure.
Pulmonary and systemic edema
Capillary hydrostatic pressure and therefore capillary fluid filtration are strongly influenced by venous pressure (click here for more details). Therefore, diuretics, by reducing blood volume and venous pressure, lower capillary hydrostatic pressure, which reduces net capillary fluid filtration and tissue edema. Because left ventricular failure can cause life-threatening pulmonary edema, most heart failure patients are treated with a loop diuretic to prevent or reduce pulmonary edema. Diuretics may also be used to treat leg edema caused by right-sided heart failure or venous insufficiency in the limb.
Specific Drugs
Specific drugs comprising the five classes of diuretics are listed in the following table. Carbonic acid inhibitors are not included.
| Class | Specific Drugs | Comments |
| Thiazide | chlorothiazide | primarily used for treating edema |
| chlorthalidone | thiazide-like in action, not structure; long half-life; more effective than hydrochlorothiazide in hypertension | |
| hydrochlorothiazide | prototypical drug | |
| indapamide | thiazide-like in action, not structure; long half-life; more effective than hydrochlorothiazide in hypertension | |
| methyclothiazide | thiazide-like in action, not structure; not available in the U.S. | |
| metolazone | thiazide-like in action, not structure; primarily used for treating edema | |
| Loop | bumetanide | short half-life (~ 1.5 hr); oral and I.V. |
| ethacrynic acid | non-sulfonamide used in patients with sulfa allergy; highest incidence of ototoxicity | |
| furosemide | short half-life (~ 1 hr) with normal kidney function; relatively low bioavailability (~50%) with high intra- and interpatient variability | |
| torsemide | slower onset but longer half-life (3-4 hr); oral and I.V. | |
| K+-sparing | amiloride | distal tubule Na+-channel inhibitor |
| eplerenone | aldosterone receptor antagonist; fewer side effects than spironolactone | |
| spironolactone | aldosterone receptor antagonist; side effect: gynecomastia | |
| triamterene | distal tubule Na+-channel inhibitor |
Adverse Side Effects and Contraindications
The most important and frequent problem with thiazide and loop diuretics is hypokalemia. This sometimes requires treatment with potassium supplements or with a potassium-sparing diuretic. A potentially serious side effect of potassium-sparing diuretics is hyperkalemia. Other side effects and drug interactions are listed below:
| Class | Adverse Side Effects | Drug Interactions |
| Thiazide |
|
|
| Loop |
|
|
| K+-sparing |
|
|
Revised 01/29/2026