 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Beta-Adrenoceptor Agonists (β-agonists)
General pharmacology
 Beta-adrenoceptor agonists (β-agonists) bind to β-receptors on cardiac and smooth muscle tissue. They also have important actions in other tissues, especially bronchial smooth muscle (relaxation), the liver (stimulate glycogenolysis) and kidneys (stimulate renin release). Beta-adrenoceptors normally bind to norepinephrine released by sympathetic adrenergic nerves, and to circulating epinephrine. Therefore, β-agonists mimic the actions of sympathetic adrenergic stimulation acting through β-adrenoceptors. Overall, the effect of β-agonists is cardiac stimulation (increased heart rate, contractility, conduction velocity, relaxation) and systemic vasodilation. Arterial pressure may increase, but not necessarily because the fall in systemic vascular resistance offsets the increase in cardiac output. Therefore, the effect on arterial pressure depends on the relative influence on cardiac versus vascular β-adrenoceptors. Long-term exposure to β-agonists can cause β-receptor down-regulation, which limits their therapeutic efficacy to short-term application. Beta-agonists, because they are catecholamines, have a low bioavailability and therefore must be given by intravenous infusion.
Beta-adrenoceptor agonists (β-agonists) bind to β-receptors on cardiac and smooth muscle tissue. They also have important actions in other tissues, especially bronchial smooth muscle (relaxation), the liver (stimulate glycogenolysis) and kidneys (stimulate renin release). Beta-adrenoceptors normally bind to norepinephrine released by sympathetic adrenergic nerves, and to circulating epinephrine. Therefore, β-agonists mimic the actions of sympathetic adrenergic stimulation acting through β-adrenoceptors. Overall, the effect of β-agonists is cardiac stimulation (increased heart rate, contractility, conduction velocity, relaxation) and systemic vasodilation. Arterial pressure may increase, but not necessarily because the fall in systemic vascular resistance offsets the increase in cardiac output. Therefore, the effect on arterial pressure depends on the relative influence on cardiac versus vascular β-adrenoceptors. Long-term exposure to β-agonists can cause β-receptor down-regulation, which limits their therapeutic efficacy to short-term application. Beta-agonists, because they are catecholamines, have a low bioavailability and therefore must be given by intravenous infusion.
Heart
Beta-agonists bind to beta-adrenoceptors in cardiac nodal tissue, the conducting system, and contracting myocytes.
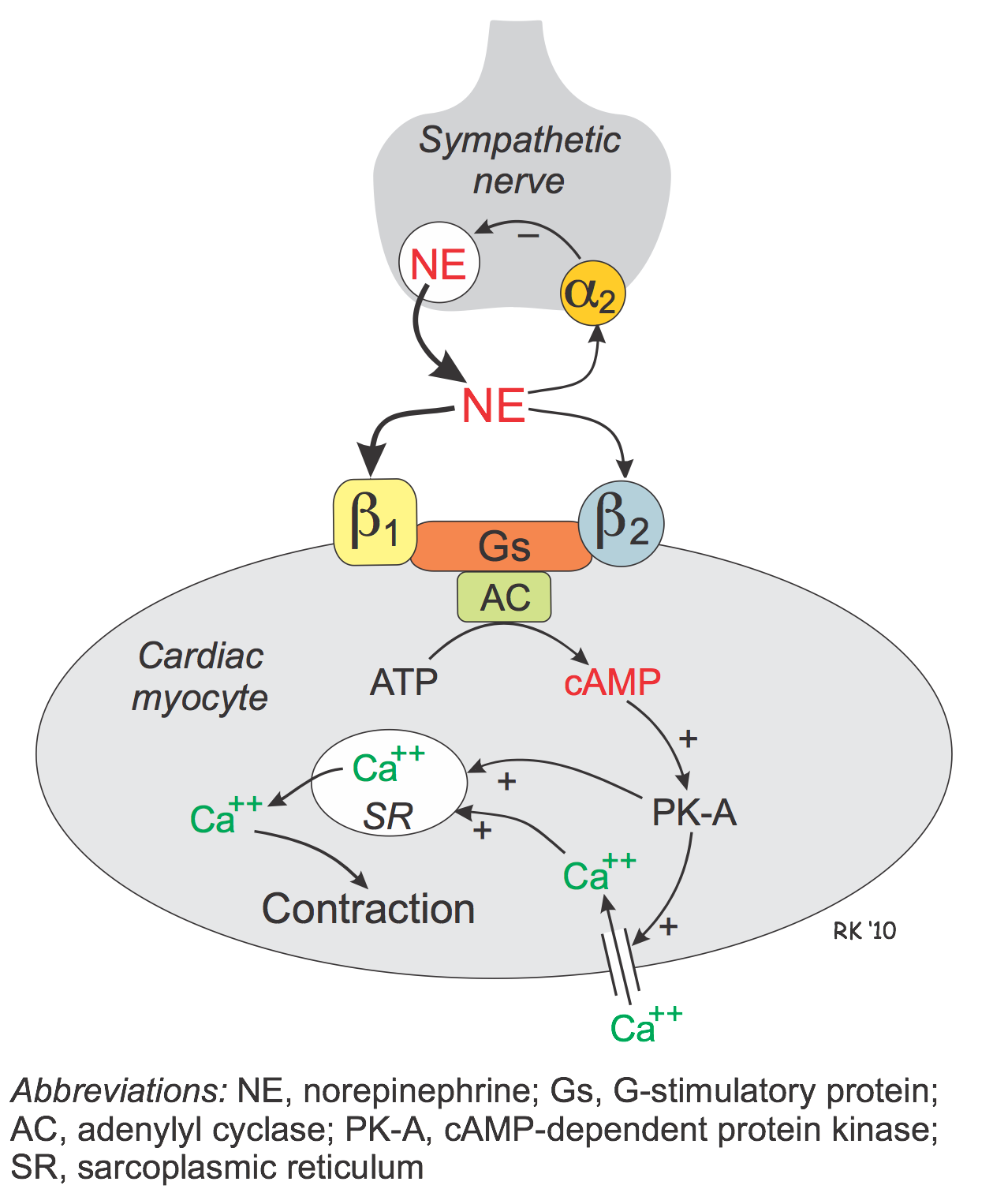
The heart has both β1 and β2 adrenoceptors, although the predominant receptor type in number and function is β1. These receptors primarily bind norepinephrine that is released from sympathetic adrenergic nerves. Additionally, they bind to norepinephrine and epinephrine that circulate in the blood.
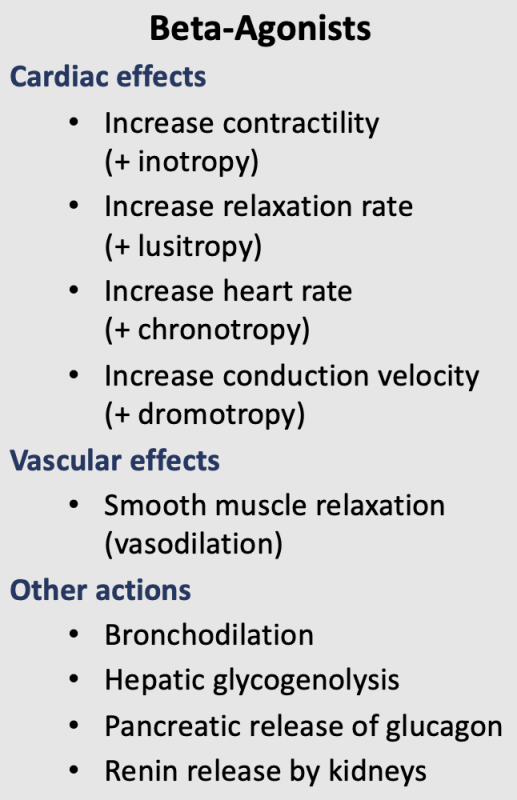 Beta-adrenoceptors are coupled to Gs-proteins, which activate adenylyl cyclase to form cAMP from ATP. Increased cAMP activates a cAMP-dependent protein kinase (PK-A) that phosphorylates L-type calcium channels, which causes increased calcium entry into the cells. Increased calcium entry during action potentials leads to enhanced release of calcium by the sarcoplasmic reticulum in the heart; these actions increase inotropy (contractility). Gs-protein activation also increases the heart rate by opening ion channels responsible for pacemaker currents in the sinoatrial node. PK-A phosphorylates sites on the sarcoplasmic reticulum, which enhances the release of calcium through the ryanodine receptors (ryanodine-sensitive, calcium-release channels) associated with the sarcoplasmic reticulum. This provides more calcium for binding the troponin-C, which enhances inotropy. Finally, PK-A can phosphorylate myosin light chains, which may also contribute to the positive inotropic effect of beta-adrenoceptor stimulation. In summary, the cardiac effects of a β-agonist are increased heart rate, contractility, conduction velocity, and relaxation rate.
Beta-adrenoceptors are coupled to Gs-proteins, which activate adenylyl cyclase to form cAMP from ATP. Increased cAMP activates a cAMP-dependent protein kinase (PK-A) that phosphorylates L-type calcium channels, which causes increased calcium entry into the cells. Increased calcium entry during action potentials leads to enhanced release of calcium by the sarcoplasmic reticulum in the heart; these actions increase inotropy (contractility). Gs-protein activation also increases the heart rate by opening ion channels responsible for pacemaker currents in the sinoatrial node. PK-A phosphorylates sites on the sarcoplasmic reticulum, which enhances the release of calcium through the ryanodine receptors (ryanodine-sensitive, calcium-release channels) associated with the sarcoplasmic reticulum. This provides more calcium for binding the troponin-C, which enhances inotropy. Finally, PK-A can phosphorylate myosin light chains, which may also contribute to the positive inotropic effect of beta-adrenoceptor stimulation. In summary, the cardiac effects of a β-agonist are increased heart rate, contractility, conduction velocity, and relaxation rate.
Blood vessels
Vascular smooth muscle has β2-adrenoceptors that have a high binding affinity for circulating epinephrine and a lower affinity for norepinephrine released by sympathetic adrenergic nerves.
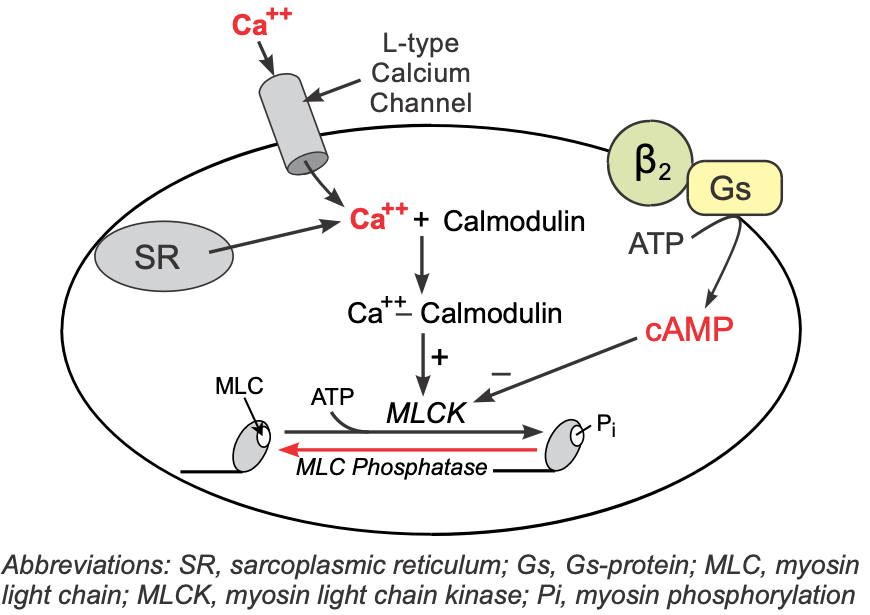 These receptors, like those in the heart, are coupled to a Gs-protein, which stimulates the formation of cAMP. Although increased cAMP enhances cardiac myocyte contraction (see above), in vascular smooth muscle, an increase in cAMP leads to smooth muscle relaxation. This is because cAMP inhibits myosin light chain kinase that is responsible for phosphorylating smooth muscle myosin. Therefore, increased intracellular cAMP caused by β2-agonists inhibits myosin light chain kinase, producing less contractile force (i.e., promoting relaxation).
These receptors, like those in the heart, are coupled to a Gs-protein, which stimulates the formation of cAMP. Although increased cAMP enhances cardiac myocyte contraction (see above), in vascular smooth muscle, an increase in cAMP leads to smooth muscle relaxation. This is because cAMP inhibits myosin light chain kinase that is responsible for phosphorylating smooth muscle myosin. Therefore, increased intracellular cAMP caused by β2-agonists inhibits myosin light chain kinase, producing less contractile force (i.e., promoting relaxation).
Other tissues
Activation of β2-adrenoceptors in the lungs causes bronchodilation. β2-adrenoceptor activation leads to hepatic glycogenolysis and pancreatic release of glucagon, which increases plasma glucose concentrations. β1-adrenoceptor stimulation in the kidneys causes the release of renin, which stimulates the production of angiotensin II and the subsequent release of aldosterone by the adrenal cortex.
Specific Drugs and Therapeutic Uses
There are several β-agonists that are used clinically to treat heart failure or circulatory shock, all of which are natural catecholamines or analogs. Most β-agonists, however, have some α-agonist activity. One agonist, dopamine, also binds to dopaminergic receptors (D1 receptors) that are found in the systemic vasculature, particularly in the kidneys, and causes vasodilation. These drugs, along with their agonist properties, are given in the table below.
| Drug | Receptor Selectivity | Clinical Use | Comments |
| Epinephrine | β1, β2 α1, α2 | Anaphylactic shock; cardiogenic shock; cardiac arrest | Low concentrations: cardiac stimulation (β1 and β2) and decreased systemic vascular resistance (↓SVR; β2); high concentrations: ↑SVR as Epi binds to α1 and α2, which increases arterial pressure. |
| Norepinephrine | β1, α1, α2 | Severe hypotension; septic shock | Increases SVR: (α1, α2) and arterial pressure; baroreflex-induced bradycardia masks direct stimulatory effects on sinoatrial node. Unlike Epi, Norepi binds weakly to β2 receptors. |
| Dopamine | D1 β1, α1, α2 | Decompensated heart failure, cardiogenic shock and acute renal failure | Precursor of norepinephrine. Low concentrations: ↓SVR (D1); intermediate concentrations: additionally stimulate the heart (β1); high concentrations: stimulate the heart (β1) and ↑SVR as α-receptors bind to the dopamine. |
| Dobutamine | β1 | Decompensated heart failure, cardiogenic shock | Primarily β1 selective agonist causing cardiac stimulation with little change in SVR; increases arterial pressure. |
| Isoproterenol | β1, β2 | Bradycardia and atrioventricular block | Non-selective β-agonist; net effect is cardiac stimulation (β1) and vasodilation (β2) with little change in pressure. |
Side Effects and Contraindications
A major side effect of β-agonists is cardiac arrhythmia. Because these drugs increase myocardial oxygen demand, they can precipitate angina in patients with coronary artery disease. Headache and tremor are also common.
Revised 01/31/2024