 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)Class I Antiarrhythmics (Sodium-Channel Blockers)
General Pharmacology
Effects on depolarization
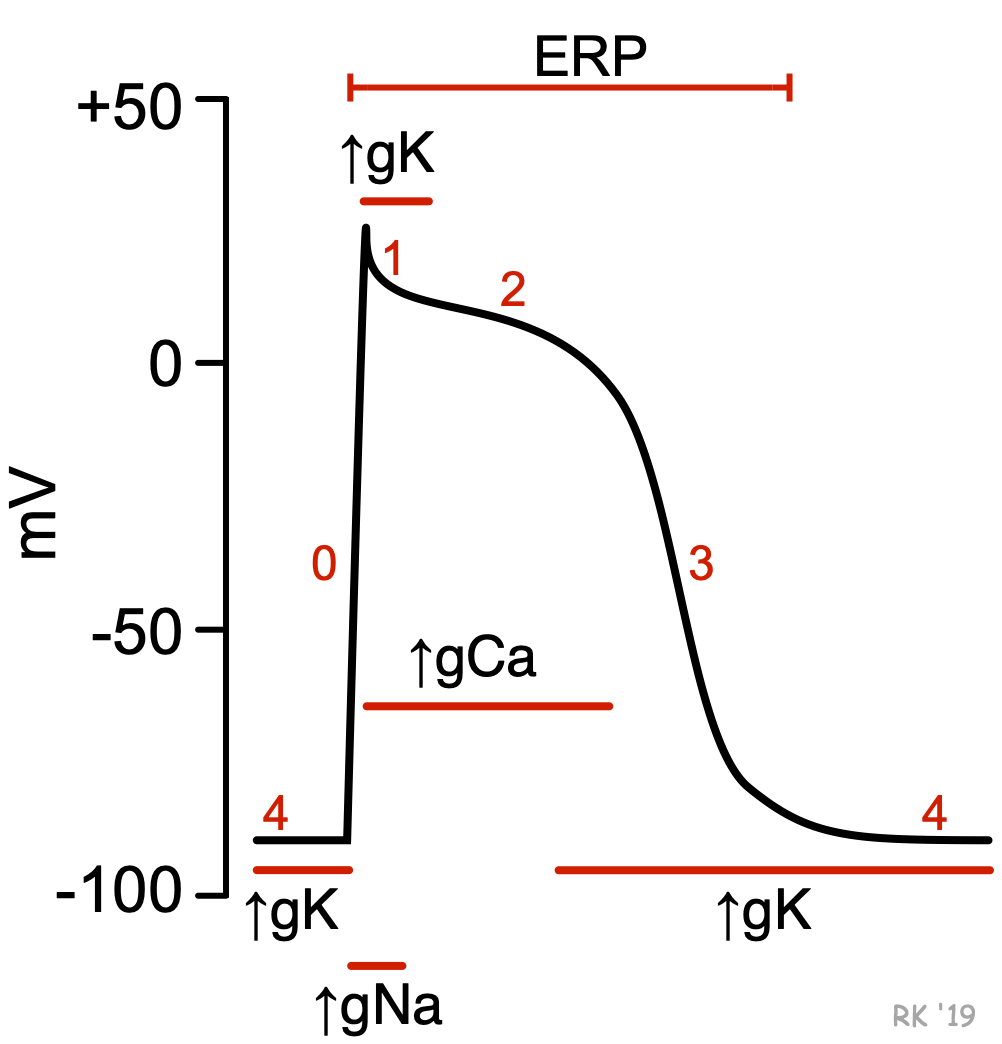 Sodium-channel blockers comprise the Class I antiarrhythmic compounds according to the Vaughan-Williams classification scheme. These drugs bind to and block fast sodium channels that are responsible for rapid depolarization (phase 0) of fast-response cardiac action potentials. This type of action potential is found in non-nodal cardiomyocytes (e.g., atrial and ventricular myocytes; Purkinje fibers). Because the slope of phase 0 depends on the activation of fast sodium-channels and the rapid entry of sodium ions into the cell (Figure: ↑gNa+), blocking these channels decreases the slope of phase 0, which also leads to a decrease in the amplitude of the action potential. In contrast, nodal tissue action potentials (sinoatrial and atrioventricular nodes) do not depend on fast sodium channels for depolarization; instead, phase 0 depolarization is carried by calcium currents. Therefore, blocking sodium channels has no direct effect on nodal tissue action potentials.
Sodium-channel blockers comprise the Class I antiarrhythmic compounds according to the Vaughan-Williams classification scheme. These drugs bind to and block fast sodium channels that are responsible for rapid depolarization (phase 0) of fast-response cardiac action potentials. This type of action potential is found in non-nodal cardiomyocytes (e.g., atrial and ventricular myocytes; Purkinje fibers). Because the slope of phase 0 depends on the activation of fast sodium-channels and the rapid entry of sodium ions into the cell (Figure: ↑gNa+), blocking these channels decreases the slope of phase 0, which also leads to a decrease in the amplitude of the action potential. In contrast, nodal tissue action potentials (sinoatrial and atrioventricular nodes) do not depend on fast sodium channels for depolarization; instead, phase 0 depolarization is carried by calcium currents. Therefore, blocking sodium channels has no direct effect on nodal tissue action potentials.
The principal effect of reducing the rate and magnitude of depolarization by blocking sodium channels is a decrease in conduction velocity in non-nodal tissue (atrial and ventricular muscle, Purkinje conducting system). The faster a cell depolarizes, the more rapidly adjacent cells become depolarized, leading to a more rapid regeneration and transmission of action potentials between cells. Therefore, blocking sodium channels reduces the velocity of action potential transmission within the heart (reduced conduction velocity; negative dromotropy). This can serve as an important mechanism for suppressing tachycardias that are caused by abnormal conduction (e.g., reentry mechanisms). By depressing abnormal conduction, reentry mechanisms can be interrupted.
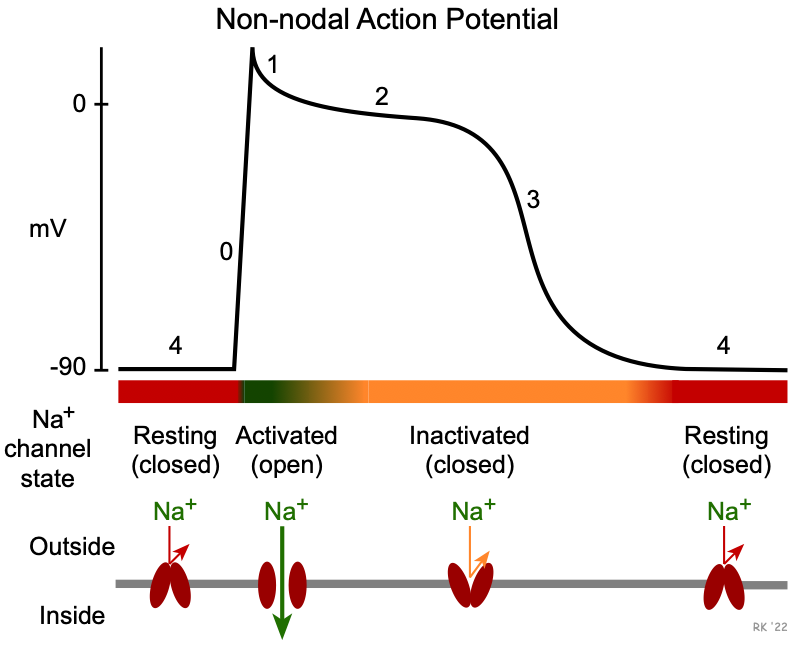 The three subclasses of sodium-channel blockers differ in how they bind to the fast sodium channel. To understand this, it is important to note that these channels have different states: resting (closed), activated (open), and inactivated (closed). These states are regulated by voltage and time-dependent mechanisms that alter conformational states of the channel structure. As shown in the figure to the right, the resting state occurs during phase 4 when the membrane potential is repolarized to its resting potential of -80 to -90 mV. When these channels are closed, inward sodium currents through these channels do not contribute to the membrane potential. When the membrane potential is rapidly depolarized to about -70 mV by an adjacent cell that becomes depolarized, these channels undergo a rapid change to the activated (open) state. This increases sodium conductance (gNa+) thereby permitting a rapid influx of sodium that further depolarizes the cell (phase 0). Once the cell depolarizes, this leads to inactivation and the closing of these channels. The many sodium channels in a single cell respond at slightly different voltages and times, so the process of inactivation, while rapid, is not instantaneous but is still occurring in early phase 2. Some of these more slowly inactivated channels produce late sodium currents. When the channels are in the inactivated (closed) state, inward sodium currents through these channels cease. As the membrane potential repolarizes during phase 3, the inactivated state prevents reactivation of action potentials (i.e., the cell is refractory). As the cell potential reaches its resting value at the end of phase 3, the sodium channels transition back into their resting (close) state, and the cell can once again be stimulated to produce a new action potential.
The three subclasses of sodium-channel blockers differ in how they bind to the fast sodium channel. To understand this, it is important to note that these channels have different states: resting (closed), activated (open), and inactivated (closed). These states are regulated by voltage and time-dependent mechanisms that alter conformational states of the channel structure. As shown in the figure to the right, the resting state occurs during phase 4 when the membrane potential is repolarized to its resting potential of -80 to -90 mV. When these channels are closed, inward sodium currents through these channels do not contribute to the membrane potential. When the membrane potential is rapidly depolarized to about -70 mV by an adjacent cell that becomes depolarized, these channels undergo a rapid change to the activated (open) state. This increases sodium conductance (gNa+) thereby permitting a rapid influx of sodium that further depolarizes the cell (phase 0). Once the cell depolarizes, this leads to inactivation and the closing of these channels. The many sodium channels in a single cell respond at slightly different voltages and times, so the process of inactivation, while rapid, is not instantaneous but is still occurring in early phase 2. Some of these more slowly inactivated channels produce late sodium currents. When the channels are in the inactivated (closed) state, inward sodium currents through these channels cease. As the membrane potential repolarizes during phase 3, the inactivated state prevents reactivation of action potentials (i.e., the cell is refractory). As the cell potential reaches its resting value at the end of phase 3, the sodium channels transition back into their resting (close) state, and the cell can once again be stimulated to produce a new action potential.
Class IA and IC drugs bind to (and therefore block) sodium channels in both activated and inactivated states, which makes them particularly effective in treating tachyarrhythmias. This is referred to as a use-dependence or state-dependence attribute of these drugs. At higher rates of cell depolarizations, the relative time the channels spend in a rested (closed) state is reduced; therefore, there is a higher probability that the drug will bind to the activated and inactivated states at higher heart rates. In contrast, Class IB drugs bind primarily to channels in the inactivated state. This characteristic of IB drugs is important because these drugs are very useful for arrhythmias in ischemic myocardium. This is because ischemia leads to slow cellular depolarization that inactivates sodium channels, and therefore enhanced binding of IB drugs.
Effects on repolarization
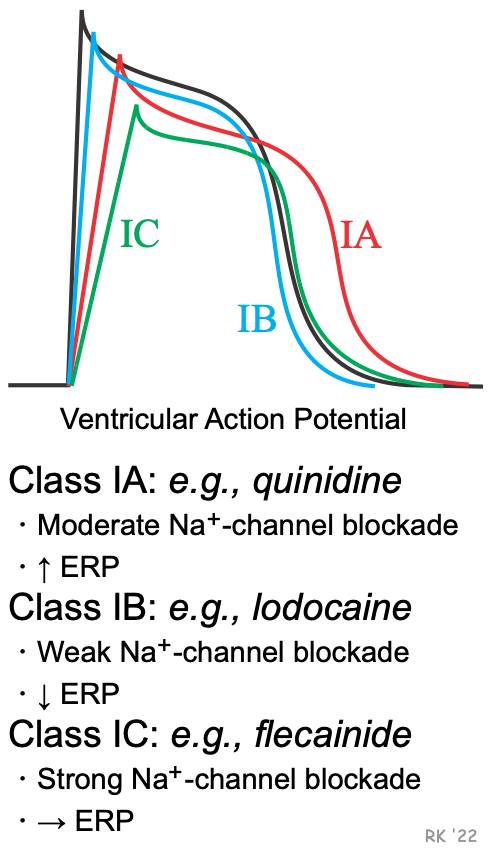 Besides affecting phase 0 of action potentials, sodium-channel blockers may also alter the action potential duration (APD) and the effective refractory period (ERP). Because some sodium-channel blockers increase the ERP (Class IA), while others decrease the ERP (Class IB) or have no effect on ERP (Class IC), the Vaughan-Williams classification recognizes these differences as subclasses of Class I antiarrhythmic drugs. These effects on ERP are not directly related to sodium channel blockade, but are related to drug actions on potassium channels involved in phase 3 repolarization of action potentials. These channels regulate potassium efflux from the cell (gK), and therefore repolarization. For example, delaying and inhibiting the activation of these outward K+ currents increases APD and ERP. The drugs in these subclasses also differ in their efficacy for reducing the slope of phase 0, with IC drugs having the greatest and IB drugs having the smallest effect on phase 0 (IA drugs are intermediate in their effect on phase 0). The following summarizes these differences:
Besides affecting phase 0 of action potentials, sodium-channel blockers may also alter the action potential duration (APD) and the effective refractory period (ERP). Because some sodium-channel blockers increase the ERP (Class IA), while others decrease the ERP (Class IB) or have no effect on ERP (Class IC), the Vaughan-Williams classification recognizes these differences as subclasses of Class I antiarrhythmic drugs. These effects on ERP are not directly related to sodium channel blockade, but are related to drug actions on potassium channels involved in phase 3 repolarization of action potentials. These channels regulate potassium efflux from the cell (gK), and therefore repolarization. For example, delaying and inhibiting the activation of these outward K+ currents increases APD and ERP. The drugs in these subclasses also differ in their efficacy for reducing the slope of phase 0, with IC drugs having the greatest and IB drugs having the smallest effect on phase 0 (IA drugs are intermediate in their effect on phase 0). The following summarizes these differences:
Sodium-channel blockade: IC > IA > IB
Increasing the ERP: IA >IC > IB (decreases)
Increasing or decreasing the APD and ERP can either increase or decrease arrhythmogenesis, depending on the underlying cause of the arrhythmia. Increasing the ERP, for example, can interrupt tachycardia caused by reentry mechanisms by prolonging the duration that normal tissue is unexcitable (its refractory period). This can prevent reentry currents from re-exciting the tissue. Increasing the APD can precipitate torsades de pointes, a type of ventricular tachycardia caused by afterdepolarizations.
Ranolazine is classified as a Class ID compound because, unlike the Class I drugs, it blocks late sodium currents found during phase 2, which leads to a delay in the activation of repolarizing potassium channels in phase 3. Therefore, ranolazine increases the APD and ERP.
Effects on automaticity
By mechanisms not understood and unrelated to blocking fast sodium channels, Class I antiarrhythmics can suppress abnormal automaticity by decreasing the slope of phase 4, which is generated by pacemaker currents.
Indirect vagal effects
The direct effect of Class IA antiarrhythmic drugs on action potentials is significantly modified by their anticholinergic actions. Inhibiting vagal activity can lead to both an increase in sinoatrial rate and atrioventricular conduction, which can offset the direct effects of the drugs on these tissues. Although IA drugs may effectively depress atrial rate during flutter, they can lead to an increase in ventricular rate because of an increase in the number of impulses conducted through the atrioventricular node (anticholinergic effect). This may require concomitant treatment with a beta-blocker or calcium-channel blocker to slow AV nodal conduction. These anticholinergic actions are most prominent at the sinoatrial and atrioventricular nodes because vagal efferent nerves extensively innervate these sites. Different drugs within the IA subclass differ in their anticholinergic actions (see table below).
Specific Drugs and Therapeutic Indications
The following table summarizes Class I drugs in terms of their therapeutic use and some special or distinguishing characteristics. More detailed information on specific drugs can be found at www.rxlist.com.
| Class IA: atrial fibrillation, flutter; supraventricular & ventricular tachyarrhythmias | ||
| quinidine* | anticholinergic (moderate) | cinchonism (blurred vision, tinnitus, headache, psychosis); cramping and nausea; enhances digitalis toxicity |
| procainamide | anticholinergic (weak); relatively short half-life | lupus-like syndrome in 25-30% of patients |
| disopryamide | anticholinergic (strong) | negative inotropic effect |
| Class IB: ventricular tachyarrhythmias (VT) | ||
| lidocaine* | IV only; VT and PVCs | good efficacy in ischemic myocardium; binds primarily to inactivated Na+-channels in ischemic cells; therefore, little effect in normal cardiac cells |
| tocainide | orally active lidocaine analog | can cause pulmonary fibrosis |
| mexiletine | orally active lidocaine analog | good efficacy in ischemic myocardium |
| Class IC: life-threatening supraventricular tachyarrhythmias (SVT) and ventricular tachyarrhythmias (VT) | ||
| flecainide* | SVT | can induce life-threatening VT |
| propafenone | SVT & VT; | β-blocking and Ca++-channel blocking activity can worsen heart failure |
| moricizine | VT; IB activity | |
| Class ID: ventricular arrhythmias (premature ventricular complexes, PVC) | ||
| ranolazine | PVC | also classified and used as an antianginal |
* prototypical drug
Abbreviations: IV, intravenous; PVC, premature ventricular complex.
Side Effects and Contraindications
The anticholinergic effects of IA drugs can produce tachycardia, dry mouth, urinary retention, blurred vision, and constipation. Diarrhea, nausea, headache, and dizziness are also common side effects of many Class I drugs. Quinidine enhances digitalis toxicity, especially if hypokalemia is present. Quinidine, by delaying repolarization, can precipitate torsades de pointes (especially in patients with long-QT syndrome), a ventricular tachyarrhythmia caused by afterdepolarizations. Disopyramide is contraindicated for patients with uncompensated heart failure because of its negative inotropic actions; propafenone can also depress inotropy. IC compounds can cause an increased risk of sudden death in patients with a prior history of myocardial infarction or sustained ventricular arrhythmias.
Revised 11/30/2023