 Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021)
Cardiovascular Physiology Concepts, 3rd edition textbook, Published by Wolters Kluwer (2021) Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)
Normal and Abnormal Blood Pressure, published by Richard E. Klabunde (2013)The Pharmacologic Treatment of Myocardial Infarction
- THIS PAGE: The Pathophysiology of Myocardial Infarction - Causes and Effects
- Page 2: Rationale for Drug Therapy in Myocardial Infarction
- Page 3: Classes of Drugs Used to Treat Myocardial Infarction
The Pathophysiology of Myocardial Infarction
Myocardial infarction ("heart attack") is the irreversible damage to myocardial tissue caused by prolonged ischemia and hypoxia. This most commonly occurs when a coronary artery becomes occluded following the rupture of an atherosclerotic plaque, which then leads to the formation of a blood clot (coronary thrombosis). If a vessel becomes completely occluded, the myocardium normally supplied by that vessel will become ischemic and hypoxic. Without sufficient oxygen, the tissue dies. The damaged tissue initially consists of a necrotic core surrounded by a marginal (or border) zone that can either recover normal function or become irreversibly damaged. The hypoxic tissue within the border zone may become a site for generating arrhythmias and display abnormal contractile function. Collateral blood flow is an important determinant of infarct size and whether the border zone becomes irreversibly damaged. Infarcted tissue does not contribute to tension generation during systole, and therefore can alter ventricular systolic and diastolic function and disrupt conduction of electrical activity within the heart. After several weeks, the infarcted tissue forms a fibrotic scar. Long-term consequences include ventricular remodeling of the remaining myocardium (e.g., development of compensatory hypertrophy or dilation), ventricular failure, arrhythmias, and sudden death.
Myocardial infarction produces clinical symptoms that include intense chest pain that may radiate into the neck, jaw, or arms (i.e., referred pain), a sense of substernal heaviness, squeezing, or pressure. Other symptoms include shortness of breath (dyspnea), fatigue, fainting (syncope), nausea, sweating (diaphoresis), anxiety, sleeplessness, hypertension, or hypotension (depending in part on the extent of cardiac damage), tachycardia and arrhythmias. Clinical studies indicate that the symptoms may be different between men and women. For example, chest pain is less common in women. Instead, their most common symptoms are weakness, fatigue, and dyspnea.
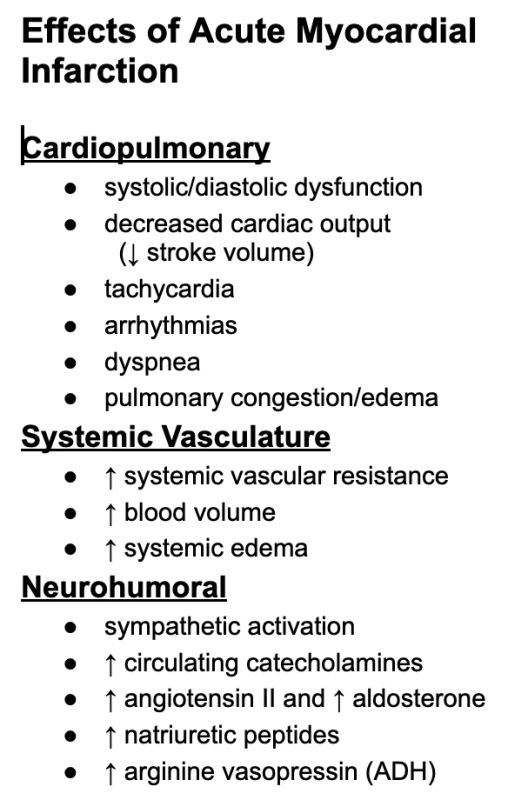 The pathophysiology of acute myocardial infarction is complex. Loss of viable myocardium impairs global cardiac function, which can lead to reduced cardiac output, and if damage is severe, to cardiogenic shock. Systolic and diastolic dysfunction are associated with ischemic myocardium. If left ventricular function is significantly impaired, pulmonary congestion and edema can occur. Ischemia can also precipitate abnormal cardiac rhythms and conduction blocks that can further impair function and become life-threatening. Reduced cardiac output and arterial pressure can elicit baroreceptor reflexes that lead to activation of neurohumoral compensatory mechanisms (e.g., activation of sympathetic nerves and the renin-angiotensin-aldosterone system) similar to what occurs during heart failure. The pain and anxiety associated with myocardial infarction further activates the sympathetic nervous system, which causes systemic vasoconstriction and cardiac stimulation (this explains why some patients become hypertensive and have tachycardia). Although sympathetic activation helps to maintain arterial pressure, it also leads to a large increase in myocardial oxygen demand that can lead to greater myocardial hypoxia, enlarge the infarcted region, precipitate arrhythmias, and further impair cardiac function. Sympathetic activation causes the diaphoresis (sweating) experienced by the patient. Renal hypoperfusion and sympathetic activation stimulate renin release, which leads to increased plasma levels of angiotensin II and aldosterone that enhance renal retention of sodium and water.
The pathophysiology of acute myocardial infarction is complex. Loss of viable myocardium impairs global cardiac function, which can lead to reduced cardiac output, and if damage is severe, to cardiogenic shock. Systolic and diastolic dysfunction are associated with ischemic myocardium. If left ventricular function is significantly impaired, pulmonary congestion and edema can occur. Ischemia can also precipitate abnormal cardiac rhythms and conduction blocks that can further impair function and become life-threatening. Reduced cardiac output and arterial pressure can elicit baroreceptor reflexes that lead to activation of neurohumoral compensatory mechanisms (e.g., activation of sympathetic nerves and the renin-angiotensin-aldosterone system) similar to what occurs during heart failure. The pain and anxiety associated with myocardial infarction further activates the sympathetic nervous system, which causes systemic vasoconstriction and cardiac stimulation (this explains why some patients become hypertensive and have tachycardia). Although sympathetic activation helps to maintain arterial pressure, it also leads to a large increase in myocardial oxygen demand that can lead to greater myocardial hypoxia, enlarge the infarcted region, precipitate arrhythmias, and further impair cardiac function. Sympathetic activation causes the diaphoresis (sweating) experienced by the patient. Renal hypoperfusion and sympathetic activation stimulate renin release, which leads to increased plasma levels of angiotensin II and aldosterone that enhance renal retention of sodium and water.
Go to Next Page
Rationale for Drug Therapy in Myocardial Infarction
Revised 11/30/2023